

Kyung-Cho Cho
Endocrine Substance Analysis Center
Significance of proteomics
Life science research has been developed around molecular biology and its central dogma, based on our understanding of the flow of genetic information from deoxyribonucleic acid (DNA), to ribonucleic acid (RNA), to protein. Genomics, the study of organisms’ genetic material, including DNA and messenger RNA (mRNA), reached a significant milestone with the completion of the ‘Human Genome Project’ in 2003, forming the foundation of life science research for decades.
However, researchers discovered that genomic data alone were inadequate for explaining phenotypes in nature. Consequently, they began focusing on proteomics, a research field aimed at elucidating the protein-related phenomena that regulate biological processes, thereby complementing genomics. Moreover, the importance of proteomics research in elucidating the etiology of diseases is increasingly being recognized.
The National Cancer Institute (NCI) in the United States has spearheaded The Cancer Genomics Atlas project, a large-scale collaborative effort to characterize genomic changes and elucidate the causes and treatments of cancer. Although various genomic changes underlying cancer have been discovered through this project, there exists a substantial gap between genomic changes and the onset and progression of cancer in many cases.
Therefore, researchers have recognized that genomic information alone is insufficient for comprehensively interpreting biological phenomena owing to various additional processes, including DNA splicing, regulation of expression, and post-translational modification (PTM) of proteins.
Proteomics research has emerged as an essential component of cancer research, including initiatives such as the Clinical Proteomic Tumor Analysis Consortium project, launched in 2011 by the NCI and the Cancer Moonshot Initiative project, a national cancer research program initiated in 2016 by the United States government. These projects aim to collect integrated genomic and proteomic cancer-related data in the field of proteogenomics, with the aim of identifying the patterns of genomic and proteomic changes that contribute most significantly to the cancer phenotype.
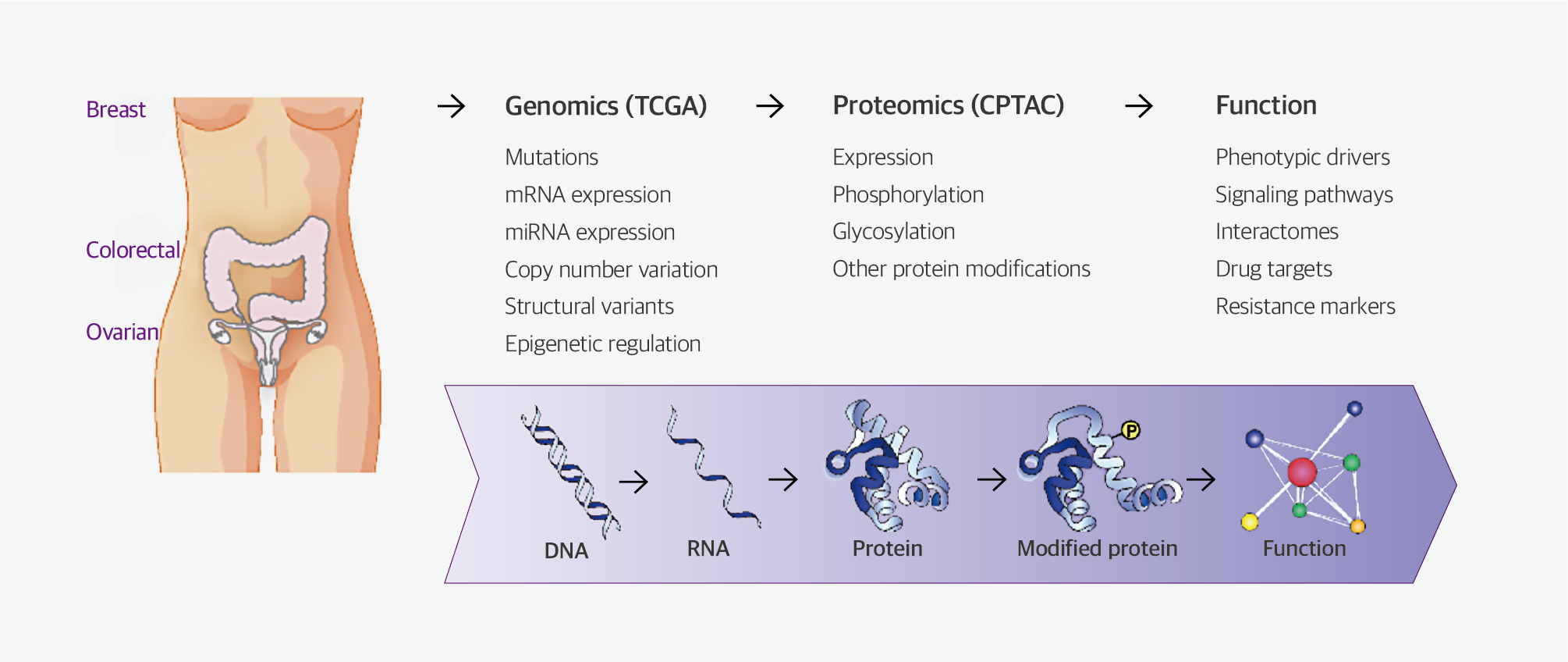
Ref) Cancer Discov. 2013 Oct;3(10):1108-12.
Fig. 1. Proteogenomic integrated workflow for the clinical proteomics tumor analysis consortium
Mass spectrometry in proteomics
A mass spectrometer is an analytical instrument capable of measuring the mass of molecules at the molecular level. Since the development of the time-of-flight (TOF) mass spectrometer in the 1940s, mass spectrometers have been widely used across various industries and institutes. These instruments became prominent in proteomics research in 1983, when Koichi Tanaka from Shimadzu Corporation in Japan successfully developed matrix-assisted laser desorption ionization (MALDI) for protein-level polymer analysis. This pioneering work earned him the Nobel Prize in Chemistry in 2002. Mass spectrometers comprise three main components: the ion source, which ionizes the molecules; the mass analyzer, which measures the mass-to-charge ratio (m/z) of the ionized molecules; and the detector, which quantifies ion abundance.
Regarding ion sources, electrospray ionization (ESI) is more efficient than MALDI for analyzing organic molecules, including proteins and peptides. Each type of mass analyzer, such as TOF or ion trap, has different strengths in terms of analysis accuracy, resolution, m/z range, and scan rate. Therefore, selecting the most suitable mass analyzer based on the analysis’ purpose is crucial.
Table 1. Types and characteristics of mass spectrometer (analyzer)
Quadrupole | Ion trap | Time of flight reflectron | Magnetic sector | FT-ICR | Q-TOF | |
Accuracy | 100 ppm | 100 ppm | < 5 ppm | < 3 ppm | < 1 ppm | < 5 ppm |
Resolution | 2’000 | 4’000 | 15’000 | 30’000 | 100’000 | 10’000 |
m/z range | 4’000 | 4-6’000 | 10’000 | 8’000 | 10’000 | 10’000 |
Scan rate | ~2 Hz | ~5 Hz | 50 Hz | 0.1 Hz | ~1 Hz | ~20 Hz |
MS/MS | MS2 (QqQ) | MSn | MS | MS2 | MSn | MS2 |
Comments | Low cost, ease of pos/neg ions switching | Low cost, ease of pos/neg ions switching | Good accuracy and resolution | High accuracy but low scan rate | High resolution & accuracy, expensive | Good accuracy and resolution |
Ref) Mass spectrometry - recent applications in chemistry, pharmacology and biology (2009)
Mass spectrometers are commonly classified according to their ionization source and mass analyzer. In proteomics research, commonly used mass spectrometers include MALDI-TOF, ESI-Q-TOF, ESI-ion trap, and ESI-FT-ICR (or Orbitrap).
Peptide sequencing for protein identification
Mass spectrometry-based proteomics offers advantages over traditional antibody-based techniques, such as enzyme-linked immunosorbent assays (ELISAs) and western blot tests, as it enables accurate identification of multiple proteins simultaneously, even in complex samples. The mass spectrometry–based protein identification process involves five main steps: (1) protein extraction, (2) peptide generation, (3) peptide separation and ionization using chromatography, (4) primary mass spectrometry analysis, and (5) tandem mass spectrometry analysis followed by database comparison.
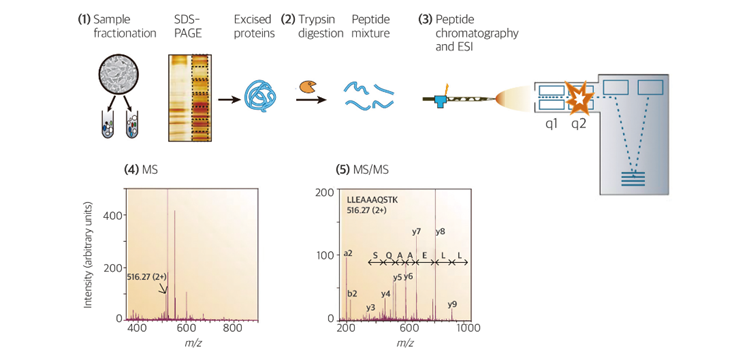
Ref) Nature. 2003 Mar 13;422(6928):198-207.
Fig. 2. Generic mass spectrometry (MS)-based proteomics experiment
In brief, proteins in biological samples undergo enzymatic digestion into peptides, typically facilitated by enzymes such as trypsin. To enhance enzyme activity, disulfide bonds between cysteine residues within proteins are reduced and alkylated. Prepared peptides are subsequently separated based on their hydrophobicity using liquid chromatography coupled with a C18 column and then sequentially eluted into the mass spectrometer. The mass analyzer is used to measure the mass of eluted peptides in primary mass spectrometry analysis, after which each peptide is fragmented at various energies in tandem mass spectrometry analysis. The mass values obtained from tandem mass spectrometry analysis are then compared to theoretical mass values of peptides in a database to identify the most similar candidates. Finally, protein identification is achieved by comparing the candidate peptides to information in the database.
Quantitative proteomics using mass spectrometry
Proteomics-based quantitative analysis can be classified into labeled quantification and label-free quantification methods, which are described in the following sections.
1. Labeled quantification
Labeled quantification methods typically involve labeling proteins or peptides with isotopically labeled compounds, differing in mass, and then comparing them to unlabeled control samples for relative quantification of proteins.
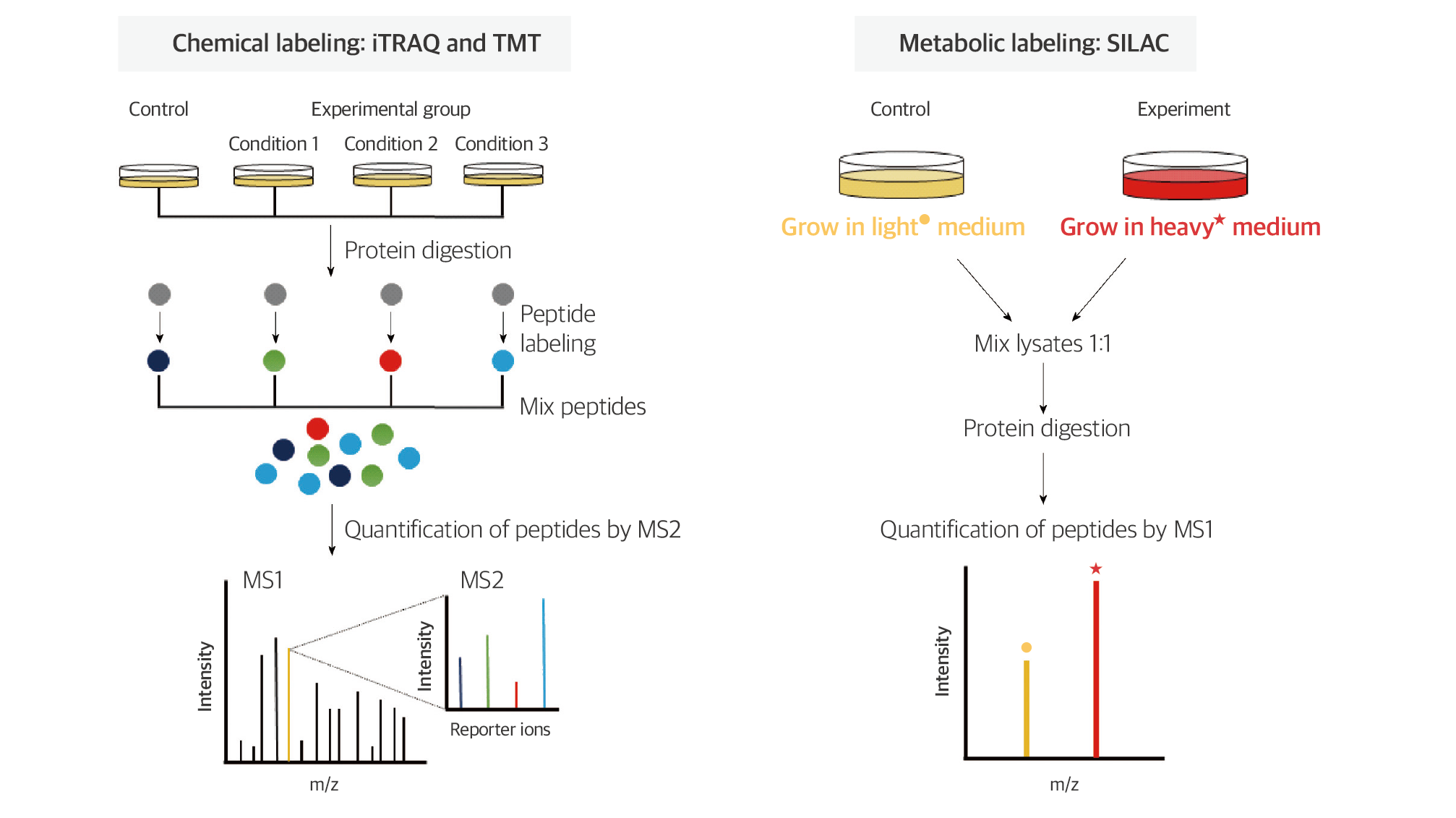
Ref) World J Gastroenterol. 2016 Oct 7;22(37):8283-8293.
Fig. 3. Quantitative proteomic strategy; labeled quantification
Among the most commonly used labeling compounds are isobaric tags for relative and absolute quantification, commonly known as iTRAQ, composed of three main components: the reporter, balance, and reactive groups. This reagent enables the analysis of 4–8 samples. During tandem mass spectrometry analysis, the intensities of reporter ions with different masses are compared, allowing relative quantification between samples.
In contrast to chemical labeling, metabolic labeling involves using media containing isotopically labeled amino acids, which replace the natural amino acids in cell culture. The stable isotope labeling with amino acids in cell culture method, commonly known as SILAC, uses media containing isotopically labeled lysine or arginine. As cells grow, the lysine and arginine within cellular proteins are substituted with isotopically labeled amino acids. By labeling samples with isotopically distinct media under different conditions, relative comparison and quantification can be achieved.
The labeled quantification method, benefiting from the ability to distinguish samples with different masses of labeling compounds, allows for the mixing and analysis of samples. This not only reduces analysis time but also ensures high quantitative accuracy, as analysis of all samples is conducted under identical conditions. Therefore, labeled quantification methods are widely used in proteomics research.
2. Label-free quantification
Label-free quantification has historically been associated with poor reproducibility and quantitative accuracy compared to labeled quantification methods. However, a mass spectrometry technique developed in recent years, namely sequential window acquisition of all theoretical mass spectra (SWATH), has emerged as a label-free quantitative analysis method capable of overcoming these limitations.
Traditionally, in mass spectrometry analysis of peptides eluted from liquid chromatography, the data-dependent acquisition method has been preferred, wherein only peptides with the highest signals from primary mass spectrometry analysis undergo tandem mass spectrometry owing to the complexity of peptide samples. With the improved performance of high-resolution mass spectrometers, it has become feasible to separate all peptides based on mass, even when analyzing complex peptide mixtures simultaneously. This advancement has facilitated the adoption of data-independent acquisition (DIA) methods, such as SWATH, which consistently obtain tandem mass spectrometry data, regardless of primary mass spectrometry results. SWATH uses complex data obtained from DIA to accurately identify proteins by comparing them with known tandem mass spectrometry data.
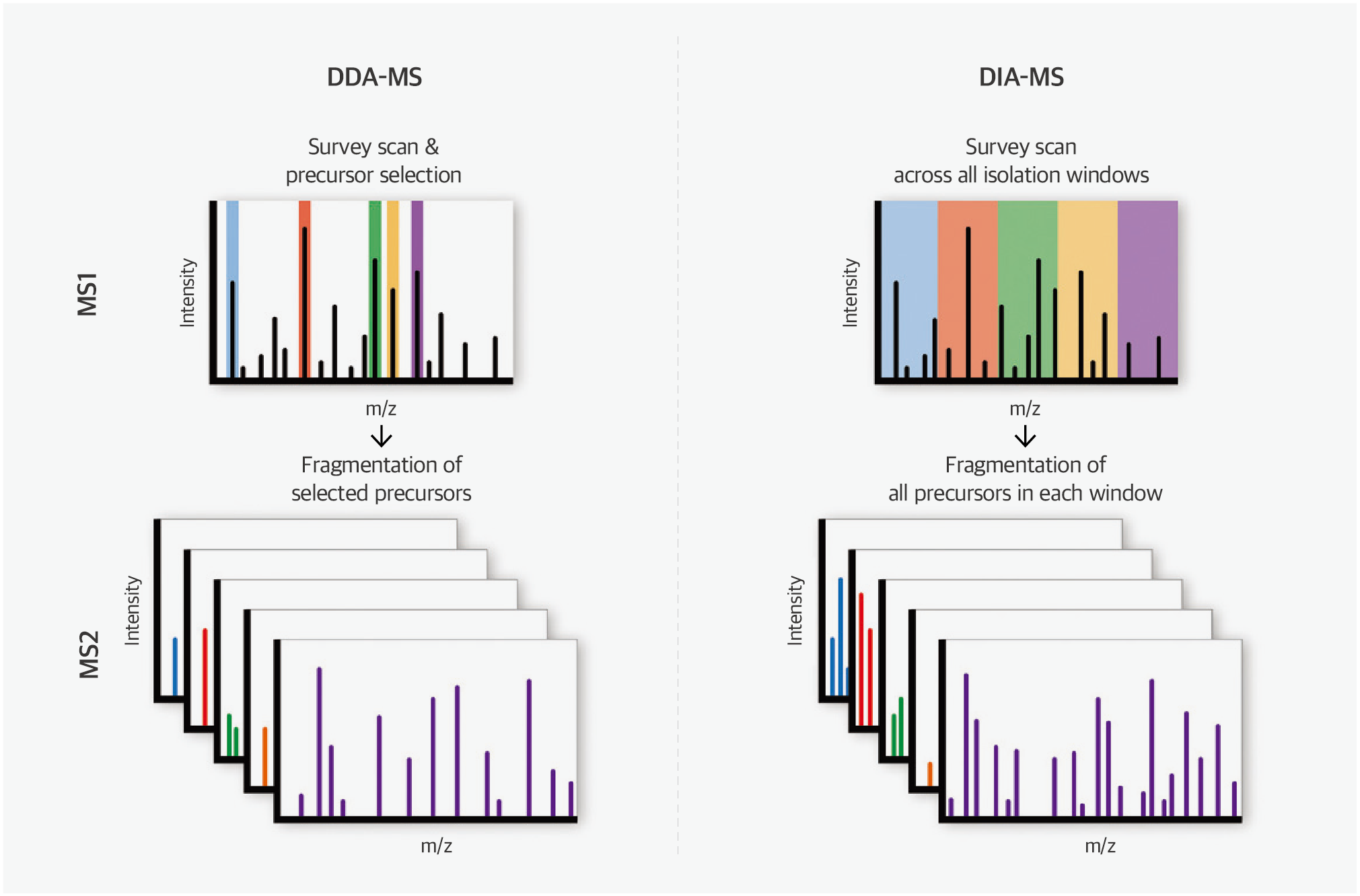
Ref) Mol Omics. 2021 Feb 1;17(1):29-42.
Fig. 4. Quantitative proteomic strategy; label free quantification
This method offers several advantages, including shorter analysis time compared with traditional methods and the ability to perform data normalization during postprocessing, thereby improving quantitative accuracy. Furthermore, label-free quantification methods markedly reduce sample preprocessing time and costs compared with labeled quantification methods, making SWATH-based label-free quantification analysis a valuable technique for large-scale sample analysis, such as clinical sample analysis.
PTM analysis of protein
Proteins undergo enzymatic or covalent modifications, known as protein PTM processes, following the translational process from mRNA. These modifications induce substantial changes in proteins’ functional activities. Among PTMs, phosphorylation within proteins is one of the most common modifications. Kinases, types of enzymes, regulate signal transduction, and cellular metabolism by controlling the phosphorylation of key proteins within the cell. Therefore, dysregulation of phosphorylation is recognized as a major cause of diseases, including cancer.
Mass spectrometry–based PTM analysis, encompassing phosphorylation, overcomes the limitations of traditional antibody-based PTM analysis, such as western blot and ELISA, which are constrained by the limited number of proteins they can analyze and the challenge in accurately determining phosphorylation sites. Therefore, mass spectrometry–based analysis is considered the most efficient PTM analysis method.
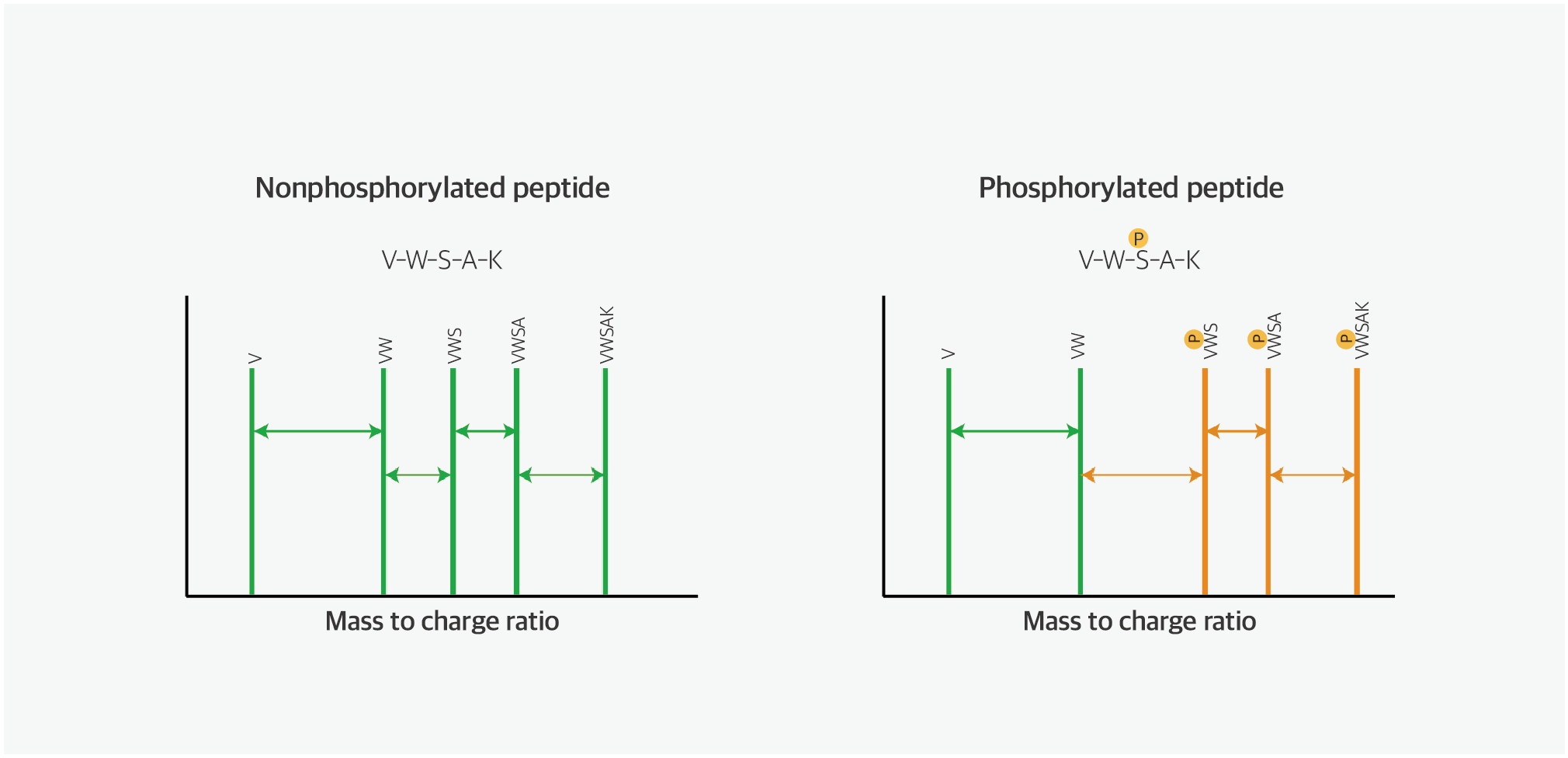
Ref) Prog Mol Biol Transl Sci. 2012:106:3-32.
Fig. 5. Phosphorylated peptide analysis using mass spectrometer
Additionally, mass spectrometry–based PTM analysis can be applied
across various fields, enabling the analysis of crucial PTMs in cell signaling,
such as methylation, acetylation, and ubiquitination, as well exploration of drug–protein
interactions.
Conclusions and perspectives
Proteomics has evolved through the advancement of technologies, such as mass spectrometry, sample preprocessing, high-resolution liquid chromatography analysis, and large-scale data processing. Although it may not have been easily accessible technology to date, proteomics has progressed from its origins as a complementary discipline to genomics to the point where data on approximately 10,000–15,000 proteins derived from nearly 20,000 genomes can be validated. Furthermore, it offers insights into PTMs and protein expression data based on tissue localization, enabling the confirmation of biological phenomena at the protein level that cannot be identified through genomics alone.
In the future, with the advancement of personalized medicine, there will be an increasing demand for individualized information on proteomes. In response to such demands, rapid developments in high-throughput technology are occurring in the field of proteomics. Technologies, including SWATH as a label-free quantification method, allow for automation from sample preprocessing to analysis. Many companies are adopting these methods; therefore, within the next few years, such services may become readily available, similar to next-generation sequencing services.
Reference
01. Ellis MJ, Gillette M, Carr SA, Paulovich AG, Smith RD,
Rodland KK, et al. Clinical Proteomic Tumor Analysis
Consortium (CPTAC).
Connecting genomic alterations to cancer biology with proteomics: the NCI
Clinical
Proteomic Tumor Analysis Consortium. Cancer Discov. 2013
Oct;3(10):1108-12.
02. Lane CS. Mass spectrometry-based proteomics in the life sciences. Cell Mol Life Sci. 2005 Apr;62(7-8):848-69.
03. Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003 Mar 13;422(6928):198-207.
04. Kang C, Lee Y, Lee JE. Recent advances in mass
spectrometry-based proteomics of gastric cancer. World J
Gastroenterol. 2016
Oct 7;22(37):8283-8293.
05. Krasny L, Huang PH. Data-independent acquisition mass
spectrometry (DIA-MS) for proteomic applications in
oncology. Mol Omics. 2021
Feb 1;17(1):29-42.
06. St-Denis N, Gingras AC. Mass spectrometric tools for
systematic analysis of protein phosphorylation. Prog Mol
Biol Transl Sci.
2012;106:3-32.