

Chang Ahn Seol, M.D.
Department of Laboratory Medicine
Importance of CNV in NGS Panels
The primary purpose of NGS panel testing for hereditary diseases is to detect nucleotide sequence variants in target genes. However, with the advancement of bioinformatics analysis techniques and the inclusion of a sufficient number of samples within a single batch, it is now possible to detect copy number variants such as deletions or duplications in gene regions by statistically analyzing sequencing depth data. Currently, such analysis pipelines have been commercialized, allowing CNV analysis to be actively conducted even in clinical laboratories performing NGS tests.
According to the literature on CNV analysis using targeted NGS panels, CNVs are detected in about 1~2% of all NGS panel tests. However, among the cases reported as positive due to pathogenic variant, CNVs account for a higher proportion, approximately 9~10%. The distribution of reported CNVs also varies depending on the type of panel used; notably, neurogenetic disease panels show a high reporting rate of over 20~30%. Thus, CNV analysis in NGS tests provides useful complementary information, especially in diagnostically challenging cases.
Ref) https://www.pacb.com/
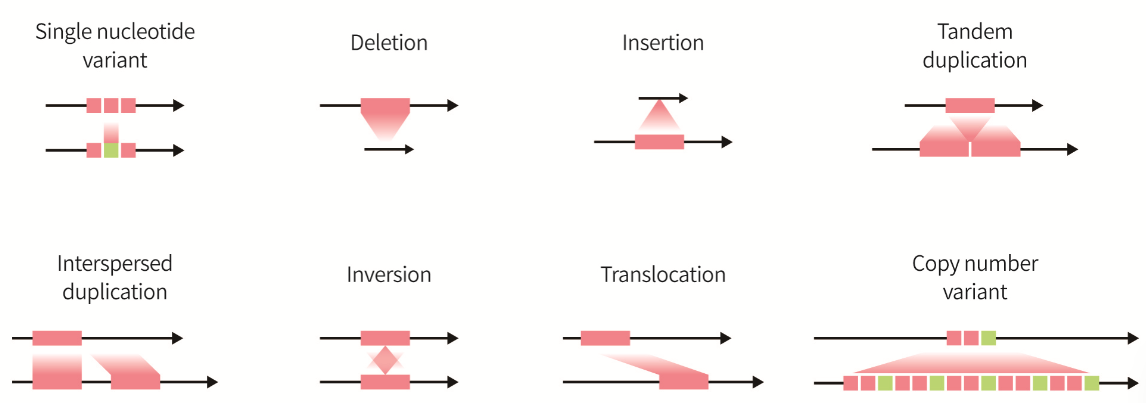
Fig. 1. Types of genomic variants
The Importance of CNV in NGS Panels
1. Statistical Data Analysis Methods for Target Gene Region
If there are 10 or more samples in a batch, deletions
or duplications in the coding regions of target genes can be identified through
statistical analysis. Representative statistical indicators include z-score and
z-ratio. Algorithms such as VisCap are frequently used
in NGS panels.
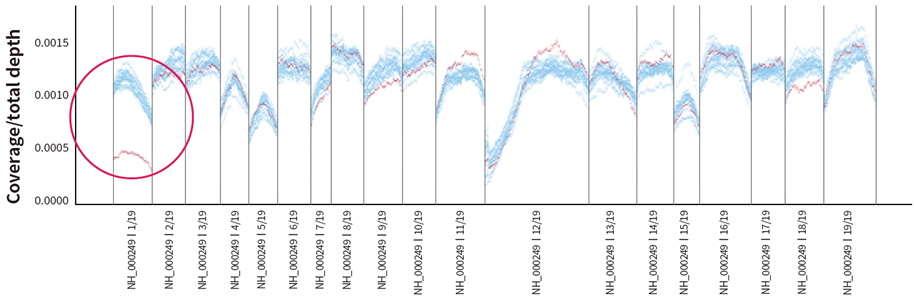
Fig. 2. Deletion of exon 1 in MLH1 gene detected in hereditary cancer panel
2. CNV Detection Method in WES (whole exome sequencing)
Most hereditary disease NGS panel tests are now
conducted in the form of WES, making it possible to perform CNV analysis based
on WES, even for panel tests. In WES testing, commonly used algorithms include
XHMM, CODEX, Conifer, etc. The results of these analyses can be visualized
using z-score and z-ratio plots, enabling the identification of large-scale
deletions and duplications, similar to CMA.
3. Off-target analysis
Even in targeted NGS panels or WES tests, sequencing
data from off-target regions can be utilized
for CNV analysis. This method, known as off-target CNV analysis, is
particularly useful for detecting large deletions or duplications. Additionally,
it offers the advantage of estimating breakpoints to some extent.
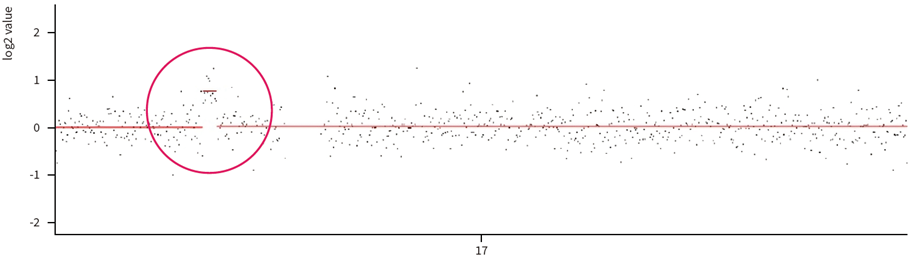
Fig. 3. Approximately 1.2 Mb duplication on chromosome 17 detected in Charcot-Marie-Tooth disease panel
(off-target analysis)
4. Analysis using IGV program
When CNV is suspected through statistical analysis as described, using
IGV program can, in some cases, allow for the identification of split reads.
This enables precise localization of deletions or duplications at the base pair
level. Additionally, though rare, it is also possible to detect genomic
rearrangement.
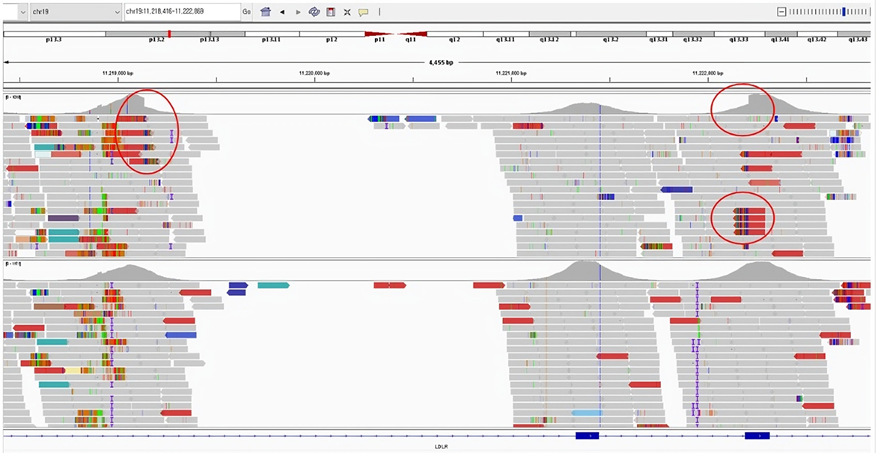
Fig. 4. Deletion findings involving exon 7 and partial exon 8 of the LDLR gene identified in the IGV program
(breakpoints can be confirmed by split read)
CNVs Frequently Detected in Hereditary Disease NGS Panels
The NGS panel with the highest detection rate of CNV is the Charcot-Marie-Tooth disease panel, identified in approximately 20% of total cases. This is because the major causative factor of this disease is a duplication in the PMP22 gene region. Additionally, other tests in which CNVs are frequently detected include panels for neurodevelopmental disorders such as autism and epilepsy, where CNVs are found in approximately 4~5% of all test cases.
In panels for short stature, skeletal dysplasia, Alport syndrome, polycystic kidney disease, and dyslipidemia, CNVs are detected in approximately 3~4% of total tests. In panels for hereditary cancer, cardiomyopathy, arrhythmia, connective tissue disease, and metabolic disorders, CNVs are detected in approximately 1~1.5% of total cases. Interestingly, in hereditary cancer and dyslipidemia panels, most reported CNVs are small, occurring at the exon level. However, in panels for autism, epilepsy, and skeletal dysplasia, large CNV—such as several hundred kilobases associated with microdeletion syndromes or over several megabases related to chromosomal imbalance translocations—are frequently reported.
Limitations
1. QC management
The primary objective of NGS panel testing is the analysis of sequence
variants. For sequence variants analysis, sufficient average coverage is
adequate. However, CNV analysis requires additional QC management. For efficiency, sequencing depth should be
uniformly distributed across all target regions. This is referred to as depth uniformity and is assessed
using various indicators, such as the coefficient of variation.
Particularly, coverage uniformity must meet a certain threshold, and values such as MAPD must be below the acceptable range. However, due to issues with specimen quality or other unknown factors, these QC values may fall outside the acceptable range. Consequently, maintaining QC for CNV analysis as reliably as with CMA is challenging, which can sometimes lead to difficulties in performing accurate CNV analysis.
2. Confirmation Testing for Detected CNV
Even if CNVs detected through NGS have been reported,
additional confirmatory testing may still be required in some cases. Confirmation
using an alternative method is recommended, particularly when CNV is suspected
in cases such as single exon deletion or in regions that are difficult to
analyze by NGS—such as homologous regions or areas with insufficient coverage. MLPA
is commonly considered, but due to limitations in probes, it cannot confirm all
genes. An alternative is a CMA-based test such as the CytoScan Xon array, which
can detect exon-level regions; however, it is currently limited to research
purposes in clinical laboratories.
Summary
In current NGS-based genetic testing, CNV analysis is often performed concurrently, as CNVs play a significant role in genetic diagnosis and contribute substantially to improving diagnostic yield. Most NGS panels for hereditary diseases are performed using WES, and CNV analysis based on WES data is conducted with increasing precision and diversity. Notably, CNVs can be detected across a broad range, from the single exon level to Mb-scale large deletion. When a pathogenic CNV associated with a disease is detected, it is reported as a positive. In particular, accurate interpretation and reporting of CNV—supported by the interpreting specialist’s extensive clinical experience and analytical expertise—greatly enhance the diagnostic accuracy for hereditary diseases.
References
01. Truty R, Paul J, Kennemer M, et al. Prevalence and properties of intragenic copy-number variation in
Medelian disease genes. Genet Med. 2019;21:114-23.
02. Mancini-DiNardo D, Judkins T, Kidd J, et al. Detection of large rearrangements in a hereditary pan-
cancer panel using next-generation sequencing. BMC Med Genomics. 2019;12(1):138.
03. Raca G, Astbury C, Behlmann A, et al. Points to consider in the detection of germline structural
variants using next-generation sequencing: A statement of the American College of Medical Genetics
and Genomics (ACMG). Genet med. 2023;25(2):100316.